Information for patients and caregivers
Information on STXBP1-E for patients and caregivers. NEW: find links to other STXBP1-E related content and social communities!
STXBP1 Encephalopathy is a severe genetic disease. We, clinical and fundamental researchers at the Vrije Universiteit (VU) Amsterdam, the Netherlands, work together to better understand STXBP1-E. We aim to improve diagnosis and therapeutic interventions, and to provide (animal and cellular) models to study the disease. This website is dedicated to inform patients, caregivers, scientists, clinicians and others interested about this disease and our work. Find the most recent developments at the bottom of this page or here.
We have added Dutch translations to the text elements. They are marked by a bold N in front.
We hebben Nederlandse vertalingen toegevoegd aan de tekstelementen van deze webpagina. Ze zijn gemarkeerd met een vetgedrukte N ervoor.
N: STXBP1-Encefalopathie (STXBP1-E) is een ernstige genetische hersenaandoening. In het STXBP1-E Centrum Amsterdam, bestaande uit klinisch en fundamenteel onderzoekers van de Vrije Universiteit Amsterdam, werken wij samen om deze ziekte beter te begrijpen. We doen onderzoek naar cel- en diermodellen van STXBP1-E, met als doel om de diagnose en behandeling van STXBP1-E te verbeteren. Deze website bevat informatie voor STXBP1-E patiënten, familieleden en mantelzorgers, onderzoekers, artsen en iedereen die geïnteresseerd is in deze ziekte en ons onderzoek. Bekijk onze meest recente ontwikkelingen onderaan deze pagina of hier.
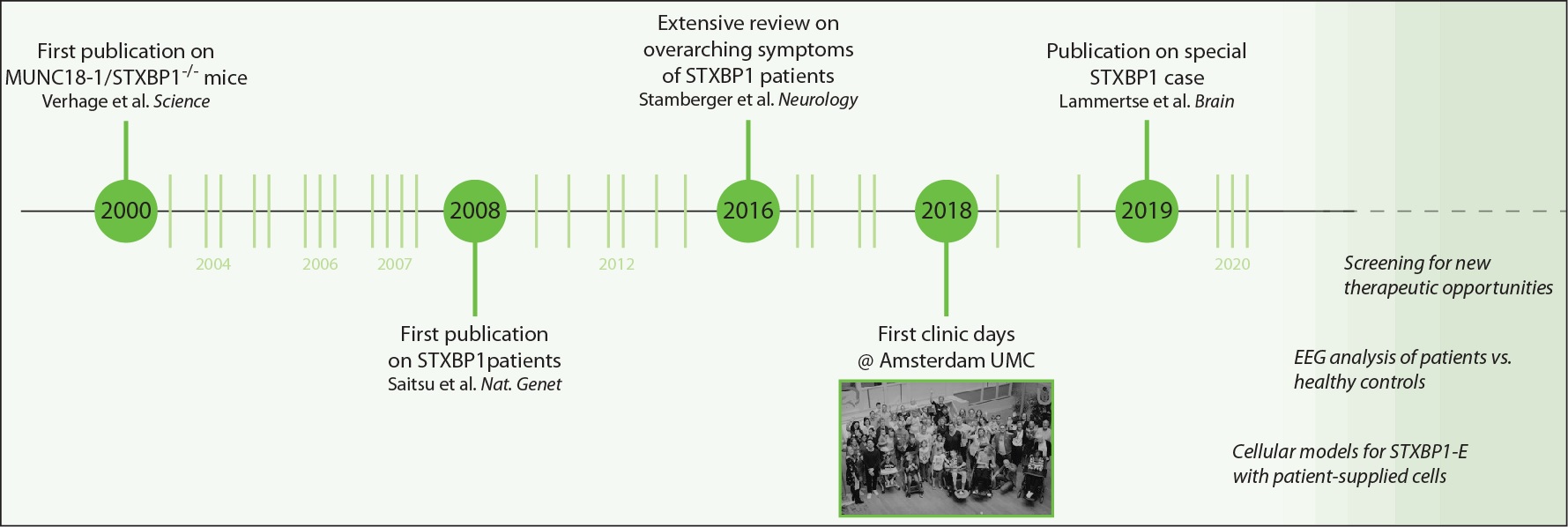
Timeline of the research development on MUNC18-1/STXBP1 since the first publication on mice lacking the protein and the discovery that it is essential for neurotransmission in 2000. In 2008, the first patients were identified.
Clinically relevant highlights in the Netherlands include the taking place of the first clinic days at the Amsterdam UMC in 2018 where many patient families came together for standardized investigations (see here and here). There are exciting times ahead with many projects in the making (more information on the bottom of this page). Light green lines represent publications on MUNC18-1 by the FGA lab over the years, which amount to more than one paper per year on average.
Last update: August 16th 2023
N: Laatste update: 16 Augustus 2023
Information on STXBP1-E for patients and caregivers. NEW: find links to other STXBP1-E related content and social communities!
Over 20 years fundamental, translational and clinical research on the STXBP1 gene and its protein MUNC18-1

How we can work together to better understand STXBP1-E

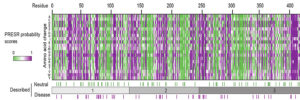
A collaboration between CNCR-FGA and University of Heidelberg shows that protein instability is the generalizable, primary cause. A new prediction tool outperforms all existing predictors. The paper is out now in Biological Psychiatry.
The STXBP1 team baked 250 cookies to raise awareness for the disorder.

De Universiteit Antwerpen en het Universitair Ziekenhuis Antwerpen doen onderzoek naar motorische problemen en gangproblemen bij kinderen en volwassenen met STXBP1-E.
